Q&A Report: Unraveling Excitation-Contraction Coupling: Simultaneous Measures of Intracellular Calcium and Action Potentials
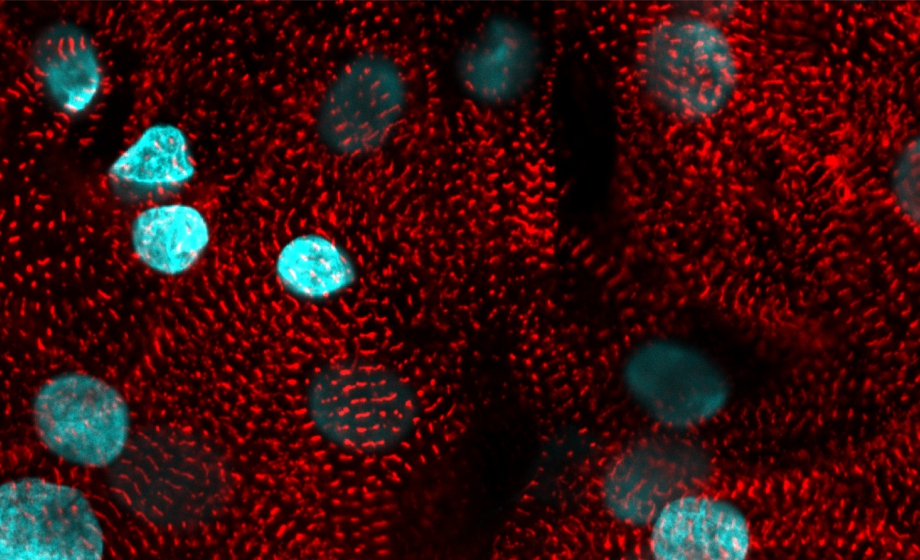
The Ca2+ transients looked quite long - are there calcium dyes with a lower Kd than Rhod-2 that will work with your combination?
The time constant of return (Tau) for the data shown is approximately 150 ms, roughly in line with human ventricular cells or other larger mammals. We zoomed in on the transients which might evoke the impression that the transients are prolonged. The kd of Rhod2 (~550nm) is higher than Fluo4(350nm) or Fura2. There are other options, X-rhod-5F (~1.7um) or X-rhod 1 (~700nm) for example both with even higher kd. Those dyes are shifted further to the red (excitation and emission) compared to rhod2, which would make a redesign of the optical setup necessary but could offer better dye separation and a broader fluovolt emission for improved SNR. Unfortunately, I am not aware of any dye with lower kd fitting the requirements for multiparametric recordings.
How long was the incubation time for verapamil?
5 min. To check how close you are to a true steady state one could periodically scan a few cells and plot the parameter of interest over time ( we store the exact time of recording within the file) to estimate when a true steady state is reached.
Why not use Fura2 free acid to record calcium transients, instead? Since this is a ratiometric dye and is more resistant to photobleaching?
Loading the cells with Fura2 would preclude the use of Fluovolt as a voltage sensitive dye, due to similarities in emission spectra. Also, Fura2 emission has a surprisingly long red tail, which poses a great challenge to combining it with available VSD of any kind.
Can Cytosolver recognize unpaced contractions for analysis or contractions with very low amplitude?
Yes it can, we can determine the start of a contraction and calculate the average beating frequency of the preparation. The analysis can be manually tweaked (setting threshold and in-/exclusion criteria). However this will result in a lower SNR and as a consequence the precision and accuracy of your results will be lower, too.
Can you explain in greater detail how your algorithm calculates contractile motion?
In IPSC derived myocytes we calculate contractions by correlating images with each other. In essence we compare the degree of similarity of a reference image (a picture taken at rest, in diastole) with all images following the reference frame. The contraction of the tissue changes the appearance and/or optical density of the tissue. Thus with higher levels of contractions, the similarity between the pictures will decrease. We express this as correlation coefficient with 1 being the perfect match and 0 reflecting no similarity between two given frames.
Can your software be used with different microscopes (not from IonOptix)?
Yes, we support most major microscopes.
Does this platform work with cardiac microtissues/3D cultures?
Yes, to a degree. The fluorescence techniques work for any preparation. The contrast-based algorithms for measuring contractility work for most preparations, but it’s best to ask IO reps to look at videos of your preps to make certain.
What filters do you use to measure with both fluovolt and rhod-2?
Excitation and emission for fluovolt are standard FITC filters, for Rhod2 we use TRITC excitation filter with an emission filter pushed a bit further to the red, the dichroic in the microscope is set up to let the 488 and 565 excitation pass while reflecting the emission of fluovolt and Rhod2.
In single cardiac myocytes, under optimal conditions, how long can you record from each cell without seeing toxic effects on action potential duration?
I managed 20 beats at 1 Hz pacing with a high light intensity without seeing obvious phototoxicity in some isolations in others it is very tricky to get 5 sec of good data. You could try to use a cocktail of antioxidants to protect the cell as described here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2957348/. We haven’t tried yet but I would definitely give it a shot.
Does the time constant of the calcium transient reflect an activation energy of tension development?
The rate limiting step during tension development always resides within the myofilaments and without direct kinetic connection between contraction and Ca2+ release I do not believe one could deduce the activation energy directly from a CaT.
Is it possible to use Cytosolver software with any equipment, like a bioteck plate reader?
The time constant of return for the data shown is approximately 150 ms, roughly in line with human ventricular cells or other larger mammals. We zoomed in on the transients which might evoke the impression that the transient are overly long. The kd of Rhod2 (~550nm) is higher than Fluo4(350nm) or Fura2. There are other options, X-rhod-5F (~1.7um) or X-rhod 1 (~700nm) for example both with even higher kd. Those dyes are shifted further to the red (excitation and emission) than rhod2, which would make a redesign of the optical setup necessary but could offer even better dye separation and a broader fluovolt emission for better SNR. Unfortunately, I am not aware of any dye with lower kd fitting the requirements for multiparametric imaging.
What equipment (allowing contraction and voltage measurements) do you recommend for iPS Cardiomyocytes that would be compatible with your software?
We test the method in adult cells, hiPSC and HL1 cells. hiPSC derived cardiac myocytes will do the job just fine as long as they contract. In general the software shown in the webinar is only compatible with our own hardware. For the measurement in hiPSC cells I would recommend the MultiCells system, the system is temperature controlled so your cells stay functional longer and the automation allows for repeated measures, which is the only way to get meaningful amplitude data from the contraction measurements in hiPSC.
Is this technology useful for the three type of measurements in isolated primary adult cardiomyocytes?
Yes, most definitely. We published a case study showing a few examples recording: https://www.ionoptix.com/multicell-case-study-demonstrates-oap-ca-cont/
What do you recommend for measurement of Ca2+ stores? Do you have any particular dye that would make able to record voltage, myoplasmic calcium and intrareticular calcium without superimposition between them? How would you load the cells? I have done Mag-Fluo-4 for this, again it was difficult.
SR Ca2+ measurements are pretty difficult indeed and combining this with myoplasmic and voltage would be quite the challenge. I am not aware of any dye combination that could work. The straight forward approach would be to swap out the Fluovolt for Fluo5N (that should be possible with the same optical setup). Measure one well with fluovolt/Rhod2 and another right after with Fluo5N/RHOD2.
If trabaculae/papillary muscle is used, can the set-up measure the cross-sectional area for force normalization?
The setup can’t measure the cross sectional area directly. The cross sectional area of trabeculae can be best approximate by an ellipsis. To get the area of such preparations you would measure the major and minor axis after dissecting the preparation to calculate the ratio. After mounting the preparation and stretching it to the desired starting length the long axis can be measured with calipers (due to the way the preparations is mounted on the setup it is tricky to get both, major and minor axis at the same time). Use the value of the major axis and calculate the minor axis based on the ratio of the un-stretched preparation.
So far we have not used papillary or trabecular muscle for multiparametric recordings. We had some success with loading such preparations with a Ca2+ dye, but no experience with Fluovolt or other voltage sensitive dyes.
Is recording of Voltage, Ca and contraction possible only in iPSC-derived cardiomyocytes?
We have tested simultaneous Ca2+, voltage and contractility measurements in adult mouse and rat myocytes, in HiPSC cells, and HL1 cells. In essence, the software used in those experiments is the same, measuring contractility in HiPSC cells only requires an additional plug-in.
We have an IonOptix system but it has a fixed view field...How do we go about getting a view field objective that could pick up various fields within one plate?
Our standard Calcium & Contractility systems come with a camera system requiring the user to align the camera along the long axis of the myocytes. For automatic image rotation as shown in the webinar, a Multicell or Multicell Light System is required.
How can we calibrate contractility when using edge detection?
Edge detection for adult cardiac myocytes can be calibrated as detailed here (page 115ff – 4.3.1 Video Calibration Dialog): https://www.ionoptix.com/resource/ionwizard-acquisition-manual/
In IPSC derived myocytes such calibration is not possible. Thus, it is not possible to compare the level of contraction from one cluster of cells with any other cluster in the same dish or other dishes/experimental days. The only way to get a meaningful data from such a setting is to perform paired measurements.
How would you reduce movement artifact from the fluorescent signal in the case that you cannot use a myosin inhibitor?
Currently we can’t account for it. In order to minimize motion artifacts we look for centers of contraction i.e. places where all cells contract towards one central point, while avoiding regions with more complex motion profiles (e.g. parts of the monolayer moves out of the ROI and other regions are pulled in at a later time form another region). Personally, I would suggest to seed cells not as dense, then you have a good chance of measuring small nests of cells and don’t need to worry about motion artifacts at all.
What's the typical concentration to be used for the fluorescent dyes?
For Calbryte 590AM we are using 8 uM (1 vial + 25 uL DMSO to dissolve + 5 mL Tyrode), and for FluoVolt we use the recommended 1uL FluoVolt + 10 uL PowerLoad per mL of Tyrode (ThermoFisher kit)
Do you recommend assessing contraction, calcium transient, and voltage in hypertrophy cardiac disease?
I recommend assessing action potential duration change with hypertrophic remodeling. There are differences depending on the species. Mostly Ito (Kv4.3, KChIP2 expression change decreases with hypertrophy) affecting AP duration in mice and notch potentials in humans.
How do you calibrate in vivo fluovolt fluorescence to membrane voltage measurements?
You cannot calibrate fluorescence and voltage. If you need absolute values you must use patch clamp. Potentiometric dyes are good for AP duration and ectopic activity.
Are you loading dyes at room temperature or at 37degC?
Room temperature
You mentioned that SERCA becomes less active; is this due to SERCA downregulation?
We measure by WB and remain unchanged. We still don’t know how it is regulated.
Could you measure in the IPSC-derived cardiomyocytes with the new protocol the changes in sarcomere length, too?
I think sarcomere length needs more brightness. There is alignment in the cardiomyocytes, but contrast is not enough to detect. Edge detection works better.
Is it possible to record Ca, Voltage, and Sarcomere length at the same time?
It is possible to measure calcium voltage and fractional shortening at the same time.
How does action potential duration regulate sarcoplasmic reticulum (SR) calcium load?
Longer action potentials keep L-type channels open for a longer time. Some papers from Dr. Donald Bers show that AP duration also regulate SR load.