Orphan Receptor GPR55: A New Therapeutic Target for Atherosclerosis?
Blog post by Nina Culum, MSc
Cardiovascular disease is the leading cause of death worldwide, representing 32% of all global deaths in 2019 [1]. One of the major contributors to this epidemic is atherosclerosis, the chronic inflammation of the arterial wall [2, 3]. Increasing evidence supports the role of T cells as drivers and modifiers of the pathogenesis of atherosclerosis [2], while clinical studies have shown that therapies that target inflammatory pathways hold potential in preventing and treating this condition [3]. Advances in analytical methodologies have led to a deeper understanding of the cellular heterogeneity of atherosclerotic lesions, which now supports the relevance of B cells in their pathophysiology [4].
Depending on their subset and activation state, B cells can exert both pro- and anti-atherogenic effects, which are mediated by their three main functions (i.e., antibody production, cytokine release, and antigen presentation) (Figure 1) [5]. Recently, antagonism of GPR55, a G protein-coupled orphan receptor that is highly expressed in splenic plasma cells and (to a lesser extent) marginal zone B cells, has been reported to have atheroprotective effects in human aortic endothelial cells [6] and has been found to increase neutrophil activation in a murine atherogenesis model [7].
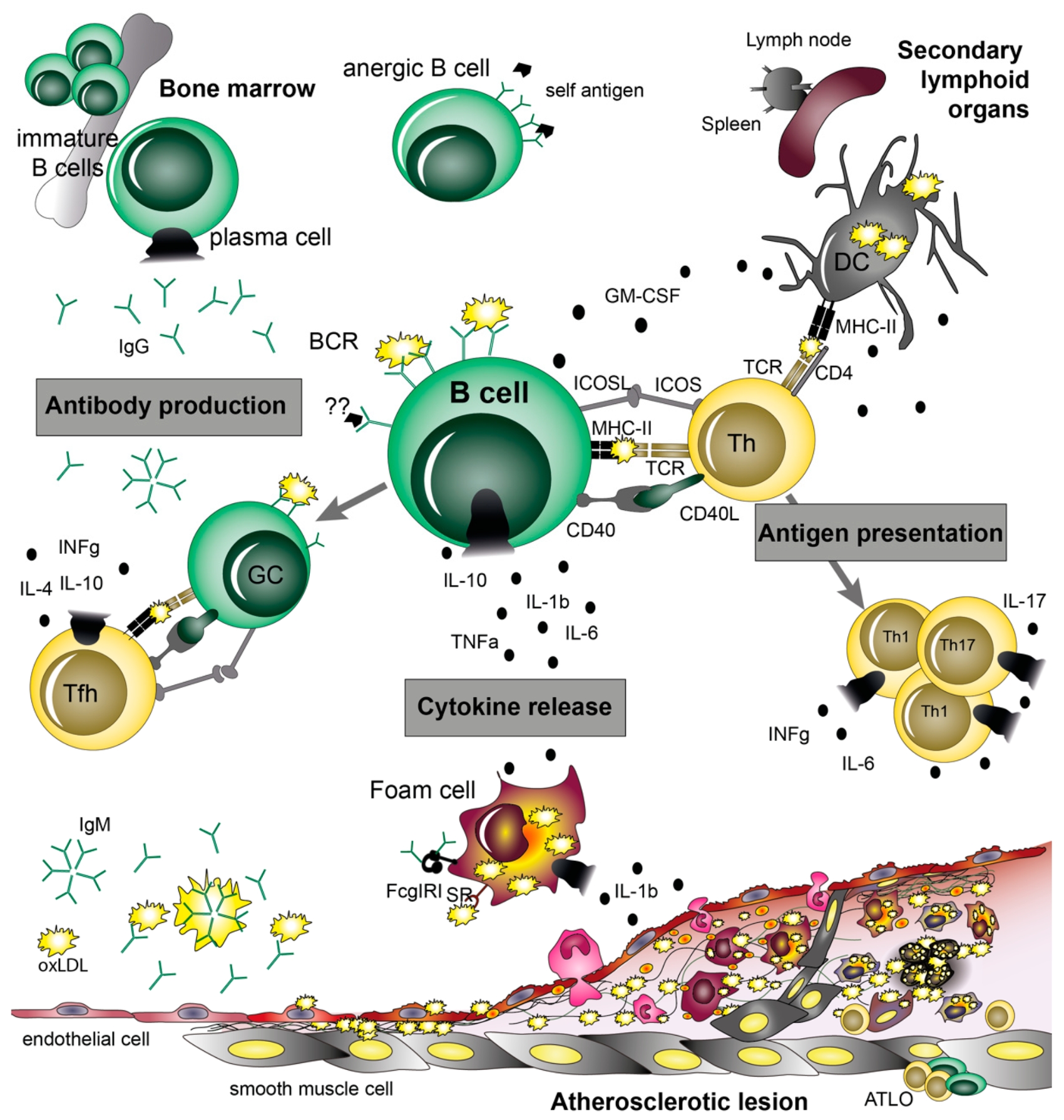
Figure 1: Illustration of B cell functions in atherosclerosis. © 2021 Ma et al., licensed under CC BY 4.0.
Since the role of GPR55 in adaptive B cell response regulation has not yet been reported in the context of atherosclerosis, Guillamat-Prats et al. have investigated how GPR55 in B cells affects atherosclerosis development [4]. In this blog post, we highlight the main findings from this study, which was published this month in Nature Cardiovascular Research.
GPR55 expression and functionality in murine and human B cells
In the first experiments described, the authors investigated the regulation and function of GPR55 signaling in atherosclerosis by feeding apolipoprotein E-deficient (Apoe-/-) mice Western diet (WD) for 4 or 16 weeks, and compared the effects to a global Gpr55 knockout (Apoe-/-Gpr55-/-) mouse model. At 4 weeks, Apoe-/-Gpr55-/- mice exhibited larger aortic plaques than Apoe-/- controls, suggesting that GPR55 signaling may counterbalance plaque development at the early disease stage. Although the difference in aortic lesion area was no longer observed at 16 weeks, the plaque burden was still found to be higher in the descending aorta of Apoe-/-Gpr55-/- mice. The authors note that these effects of Gpr55 deficiency on advanced plaque phenotype could be relevant to the complications that are observed in human atherosclerosis.
The authors also determined that Gpr55 expression was highest in B cells, followed by T cells, neutrophils, and monocytes. Additionally, the highest expression of Gpr55 was found in plasma cells, which was localized to the follicular and germinal center areas. To assess the functional GPR55 signaling response in B cells, the authors measured intracellular calcium levels in response to stimulation with the endogenous GPR55 ligand lysophosphatidylinositol (LPI). With a normal diet, increased intracellular calcium was only observed after LPI stimulation in Apoe-/- cells expressing Gpr55. Moreover, a dose-dependent calcium response to LPI was found in human peripheral blood B cells, indicating a functional LPI-GPR55 signaling pathway.

In this webinar, Dr. Michael Sturek reviews features of macrovascular atherosclerosis and microvascular dysfunction that underlie ischemic events and the need for appropriate animal models for optimal translation. WATCH NOW

Dr. Mariana Kaplan highlights the role of systemic autoimmunity in the development of vasculopathy and atherosclerosis and discusses potential strategies to prevent these complications. WATCH NOW
Effects of GPR55 deficiency in metabolism, inflammation, and atherosclerosis
Next, the authors examined how GPR55 deficiency affects metabolism, as well as vascular and systemic inflammation under hypercholesterolemic conditions. Pronounced metabolic changes such as increased body weight were observed in Apoe-/-Gpr55-/- mice compared to age-matched Apoe-/- controls, which were notable at baseline, all the way through to 16 weeks of WD. Additionally, Apoe-/-Gpr55-/- mice exhibited a hyper-inflammatory phenotype alongside increased circulating and aortic leukocyte subsets. To understand the reason behind this excessive pro-inflammatory phenotype, the authors profiled the B cell population by flow cytometry and immunohistological analysis (Figure 2). In Apoe-/-Gpr55-/- mice, B1 cells were shifted to a decreased proportion of the anti-atherogenic subset, while there was an increased proportion of the pro-atherogenic marginal zone subset.

Figure 2: Spleen sections of Apoe-/- and Apoe-/-Gpr55-/- mice stained for B cell markers. © Guillamat-Prats et al., licensed under CC BY 4.0.
Although Apoe-/-Gpr55-/- mice exhibited lower plasma cell counts, they had increased titers of immunoglobulin A (IgA) antibodies and several immunoglobulin G (IgG) subclasses. Additionally, the authors identified a positive correlation between IgG titers and plaque size in Apoe-/-Gpr55-/- mice following 4 weeks of WD, as well as elevated IgG titers even after 16 weeks of feeding. Elevated plasma levels of B cell activating factor were also found in Apoe-/-Gpr55-/- mice, which is indicative of a hyperactive B cell response during atherosclerosis in the absence of GPR55 signaling. Additionally, the authors performed bulk RNA sequencing of splenic plasma cells, blood plasmablasts, and bone marrow long-lived plasma cells from both mouse models, and confirmed that Gpr55 deficiency affects B cell maturation and plasma cell function.
To further investigate the effect of B cell-specific GPR55 signaling in atherosclerosis, the authors used a mixed bone marrow transplantation strategy with µMT as a model for mice lacking functional B cells. For these experiments, a 50/50 ratio of Gpr55-/- and µMT bone marrow was transplanted into lethally irradiated Ldlr-/- recipients, with wildtype (WT)/µMT mice used as controls. The metabolic changes that were mentioned previously were lost, with no significant differences observed in body weight between Gpr55-/-/µMT and WT/µMT mice. Additionally, Gpr55-/-/µMT mice developed larger plaques compared to controls, and had increased plasma cell counts.
Lastly, the authors injected GPR55-deficient B cells into Apoe-/- mice to further evidence the importance of functional B cell GPR55 signaling in atherosclerosis. This adoptive Gpr55-/- B cell transfer triggered an increase in plasma IgG levels and plaque size after 4 weeks of WD as compared to mice who received WT B cells, without affecting body weight or cholesterol (Figure 3).
")
Figure 3: Aortic root plaque size of B cell-depleted Apoe-/- mice receiving WT or Gpr55-/- B cell adoptive transfer and 4 weeks of WD. © Guillamat-Prats et al., licensed under CC BY 4.0.
Implications and future directions
Guillamat-Prats et al. conclude that this study provides previously unrecognized insights into the complex and critical role of GPR55 signaling in atherosclerosis, ranging from metabolic to B cell-specific effects. However, some questions remain unresolved, including the role of LPI in B cell GPR55 signaling and atherosclerosis, as well as the role of GPR55 in other immune subtypes such as T cells. Although further studies must be conducted to address these questions, GPR55 may provide a new therapeutic target to treat atherosclerosis and other chronic inflammatory diseases.
About the Author
About the Author

Nina Culum graduated from the University of Western Ontario with a Master of Science in physical and analytical chemistry. During her graduate studies, she fabricated plasmonic nanohole arrays to capture extracellular vesicles and detect cancer by surface-enhanced Raman spectroscopy. Prior to attending UWO, Nina completed her Bachelor of Science in chemistry at the University of Waterloo.
References
- World Health Organization [Internet]. Cardiovascular diseases (CVDs). 2021 Jun 11 [cited 2022 Nov 18]. Available from: www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
- Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387-401. DOI: 10.1038/s41569-020-0352-5.
- Soehnlein O, Libby P. Targeting inflammation in atherosclerosis — from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20:589-610. DOI: 10.1038/s41573-021-00198-1.
- Guillamat-Prats R, Hering D, Derle A, Rami M, Härdtner C, Santovito D, et al. GPR55 in B cells limits atherosclerosis development and regulates plasma cell maturation. Nature Cardiovasc Res. 2022;1:1056-71. DOI: 10.1038/s44161-022-00155-0.
- Ma SD, Mussbacher M, Galkina EV. Functional role of B cells in atherosclerosis. Cells. 2021;10(2):270. DOI: 10.3390/cells10020270.
- Wang Y, Pan W, Wang Y, Yin Y. The GPR55 antagonist CID16020046 protects against ox-LDL-induced inflammation in human aortic endothelial cells (HAECs). Arch Biochem Biophys. 2020;681:108254. DOI: 10.1016/j.abb.2020.108254.
- Montecucco F, Bondarenko AI, Lenglet S, Burger F, Piscitelli F, Carbone F, et al. Treatment with the GPR55 antagonist CID16020046 increases neutrophil activation in mouse atherogenesis. Thromb Haemost. 2016;116(5):987-97. DOI: 10.1160/TH16-02-0139.